24小时服务热线:19103801095
NEWS CENTER
Recommend case
contact us
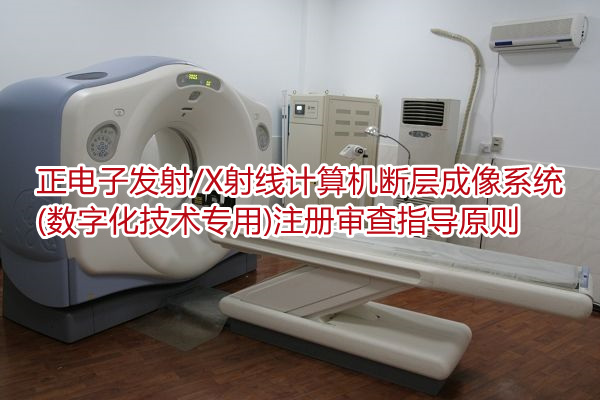
正电子发射/X射线计算机断层成像系统(数字化技术专用)注册审查指导原则 本指导原则是对正电子发射/X射线计算机断层成像系统(Imaging system of positron emission a
查看详情
CE认证是欧盟的产品安全认证,所有进入欧盟市场的医疗器械都必须进行医疗器械CE认证,医疗器械需要满足的CE指令有《有源植入性医疗器械指令》(AIMDD, 90/385/EEC)、《医疗器械指令》(MDD,93/42/EEC)和《体外诊断器械指令》(IVDD, 98/79/EC)。
查看详情
2021年12月,国家药品监督管理局共批准注册医疗器械产品211个。其中,境内第三类医疗器械产品136个,进口第三类医疗器械产品43个,进口第二类医疗器械产品29个,港澳台医疗器械产品
查看详情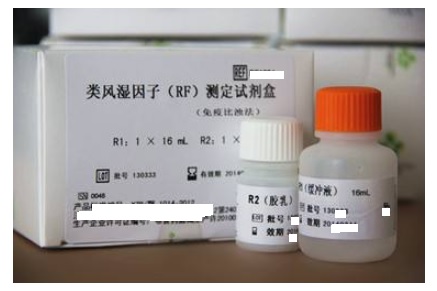
本指导原则旨在指导医疗器械注册申请人对类风湿因子检测试剂注册申报资料的准备及撰写,同时也为技术审评部门对注册申报资料的技术审评提供参考。
本指导原则是对类风湿因子测定试剂的一般要求,申请人应依据产品的具体特性确定其中内容是否适用,若不适用,需具体阐述理由及相应的科学依据,并依据产品的具体特性对注册申报资料的内容进行充实和细化。
本指导原则是供申请人和审查人员使用的指导文件,不涉及注册审批等行政事项,亦不作为法规强制执行,如有能够满足法规要求的其他方法,也可以采用,但应提供详细的研究资料和验证资料。应在遵循相关法规的前提下使用本指导原则。
本指导原则是在现行法规、标准体系及当前认知水平下制定的,随着法规、标准体系的不断完善和科学技术的不断发展,本指导原则相关内容也将适时进行调整。

本指导原则旨在指导注册申请人对特定蛋白免疫分析仪注册申报资料的准备及撰写,同时也为技术审评部门审评注册申报资料提供参考。
本指导原则是对特定蛋白免疫分析仪的一般要求,申请人应依据产品的具体特性确定其中内容是否适用,若不适用,需具体阐述理由及相应的科学依据,并依据产品的具体特性对注册申报资料的内容进行充实和细化。
本指导原则是供申请人和审查人员使用的指导文件,不涉及注册审批等行政事项,亦不作为法规强制执行,如有能够满足法规要求的其他方法,也可以采用,但应提供详细的研究资料和验证资料。应在遵循相关法规的前提下使用本指导原则。
本指导原则是在现行法规、标准体系及当前认知水平下制定的,随着法规、标准体系的不断完善和科学技术的不断发展,本指导原则相关内容也将适时进行调整。

费用是企业做经营决策最重要的考量点,没有之一。关于医疗器械注册收费标准,频繁的会有客户问到。因此,我把国内各省市医疗器械收费标准汇总做成了表格,方便大家查阅。
查看详情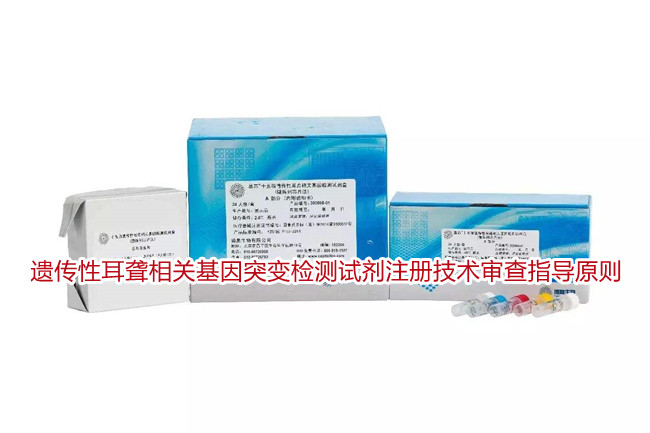
本指导原则旨在指导注册申请人对遗传性耳聋相关基因突变检测试剂注册申报资料的准备及撰写,同时也为技术审评部门对注册申报资料的技术审评提供参考。
本指导原则是对遗传性耳聋相关基因突变检测试剂的一般要求,申请人应依据产品的具体特性确定其中内容是否适用,若不适用,需具体阐述理由及相应的科学依据,并依据产品的具体特性对注册申报资料的内容进行充实和细化。
本指导原则是对申请人和审查人员的指导性文件,但不包括注册审批所涉及的行政事项,也不作为法规强制执行,如果有能够满足相关法规要求的其他方法,也可以采用,但需要提供详细的研究和验证资料,相关人员应在遵循法规的前提下使用本指导原则。
本指导原则是在现行法规和标准体系以及当前认知水平下制定的,随着法规和标准的不断完善以及科学技术的不断发展,本指导原则相关内容也将适时进行调整。

前期写了一些列文章科普医疗器械美国FDA注册流程和要求,针对I类、II类、III类医疗器械在美国的分类、注册时间、注册费用等大伙关心的热门议题做了介绍。考虑到医疗器械在美国是重监管的行业,接下来,讲下美国FDA注册注意事项。
查看详情






