24小时服务热线:19103801095
NEWS CENTER
Recommend case
contact us

医疗器械注册检验、医疗器械注册审评、医疗器械体系考核是医疗器械注册进程重大中关键路径、关键事项,本文一起来看一下医疗器械注册体系考核常见问题。
查看详情
受试者筛选是医疗器械临床试验实施阶段的先导性工作环节,筛选到合格的并且依从性很好的受试者是医疗器械临床试验的主要挑战之一,一起来了解一下。
查看详情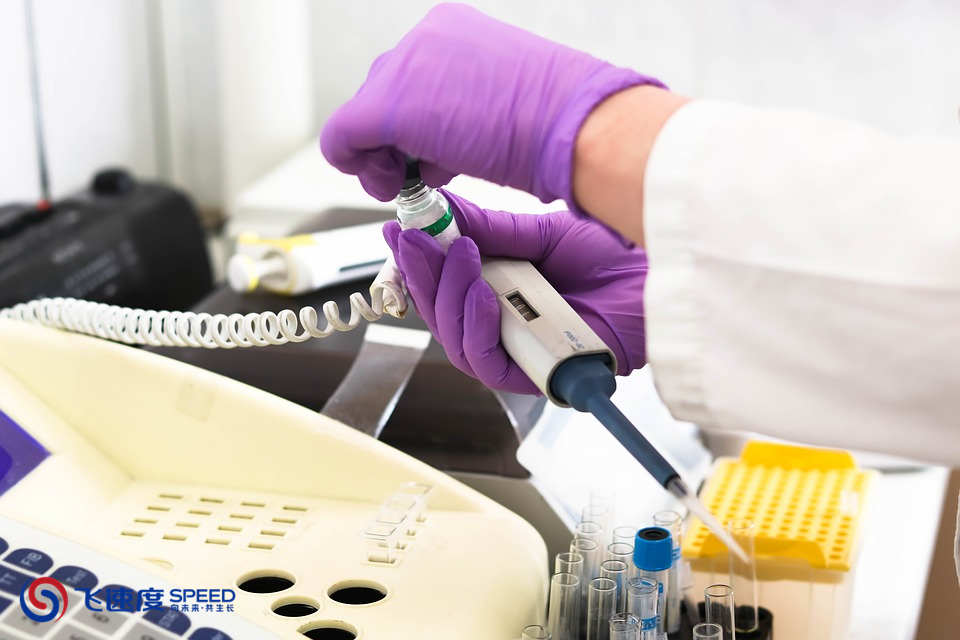
近日,药监总局最新发布18项医疗器械产品注册技术审查指导原则,详见正文。
查看详情
昨日,药监总局器审中心发布一项医疗器械注册技术审查指导原则——《笑气吸入镇痛装置注册技术审查指导原则》征求意见稿,面向大众公开征求意见。
查看详情
从美国FDA获取,首款可延缓近视隐形眼镜通过FDA医疗器械注册审批。
查看详情
刚刚,药监总局器审中心发布两条医疗器械注册技术答疑,对特定产品注册单元划分和典型型号确定提供官方解答。
查看详情
国家药监局综合司发布《医疗器械注册质量管理体系现场核查指南(征求意见稿)》,面向社会公开征求意见。
查看详情
广西药监局发布《广西壮族自治区药品监督管理局关于施行第二类医疗器械首次注册告知承诺审批的通知》,10月10日起,广西行政区域范围内企业在申报阶段可以不提交医疗器械注册检验报告!自此,企业承诺能代替某项申报资料了。
查看详情






