24小时服务热线:19103801095
NEWS CENTER
Recommend case
contact us
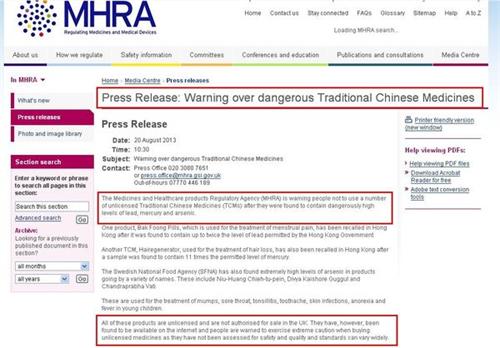
继《英国医疗器械监管白皮书》中英文版本上线后,小编从新法规中择出几点大家比较关注的问题,例如:哪些器械需要在MHRA注册?医疗器械在英国市场的注册时间和费用?相信很多小伙伴们对此类问题很感兴趣,今日就随小编一探究竟吧!
查看详情
ISO 13485标准于1996年首次出版,名为《医疗器械—质量管理体系—应用ISO 9001的具体要求》,其后于2003年改版,名为《医疗器械—质量管理体系—用于法规的要求》,最新版本为ISO 13485:2016。
查看详情
所谓医疗器械注册,是指食品药品监督管理部门根据医疗器械注册申请人的申请,按照法定程序,拟上市医疗器械的安全性、有效性研究和结果进行系统评估,以决定是否同意其申请过程。因
查看详情
昨天收到一位用户的留言:“想考一个GCP的证书,却在国家食品药品监督管理总局的网站上找不到报名的入口,求告诉哪里报名GCP考试!!能拿国家级GCP证书的,找工作用的。谢谢!!!!”
查看详情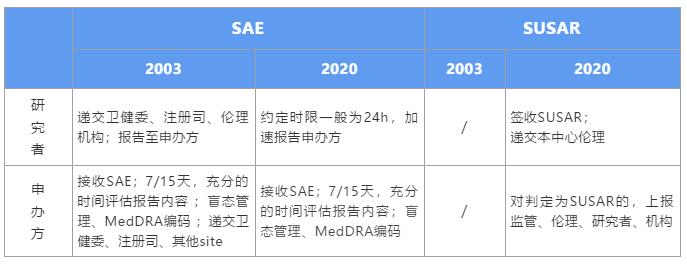
新版GCP对于申办方的影响是全方位的,就SAE个例报告处理方面,强化了申办方的主体责任,细化了申办方对SAE的管理流程。在此,我们详细解读,在新规之下,该如何高效、合规的处理SAE。
查看详情
医疗器械分为三类,一类不需要医疗器械运营企业许可证,二类、三类需要办理。要求《医疗器械经营企业许可证》必须具备以下条件: (一)有与经营规模和经营范围相适应的质
查看详情
分享一份来自德阳市人民医院的GCP培训ppt课件(文末可下载)。
查看详情
gcp证书有什么用?这个问题应该刚接触临床行业的小伙伴问的比较多,比如CRO公司的临床项目协助人员(CRA/CRC/项目经理等)、医院的临床项目团队(主要研究者/团队成员)和一些生产企业的内部人员(临床项目负责人等)都有这个证,这样看来这个证还挺重要的。下面你关心的重点来了!!!!
查看详情






