24小时服务热线:19103801095
NEWS CENTER
Recommend case
contact us

实验室原始记录用于为可追溯性提供文件、并提供验证、预防措施和纠正措施的证据,具有清晰、完整、真实和原始等特点。它们是出具检定证书,校准报告的依据。重要性不多说,总之原始记录很重要。但总有小伙伴会粗心大意,导致原始记录出错,你让他认真检查,他都不知道从哪里下手?其实他也很痛苦。。。因为,真的不知道。
查看详情
国家药监局高研院GCP培训课程,考试通过后发放国家级GCP证书,对想从事临床相关工作,入职CRO公司的你来说,证书的效力是最高的。线上报名和考试流程:官网注册账号,填写个人基础信息、报名缴费(医疗器械GCP 950元、药物GCP 1000元),然后审核通过就可以开通学习了(在线视频学习),所有学习视频看完了就可以在线考试了(25题选择+25题判断,50题×2分/题=100分),考试时长60min,医疗器械86分合格、药物82分合格,一天可以考三次,次日恢复三次机会。我第一次就过了,题目不难,时间很充足,考完第二天还是第三天证书就寄过来了。
查看详情
2021年上海市GCP证书在线培训班来了!本次GCP证书培训事宜,药学会兼具GCP证书发行资格。GCP证书是进入临床行业的敲门砖,广泛应用于CRO公司、医疗机构和各大生产企业。
查看详情
医疗器械在产品注册方面,会相对来说比较复杂一些。由于产品涉及到人使用,在安全系数方面就会格外重视。对于二类医疗器械注册,小编想和大家具体的讲一下。重庆第二类医疗器械
查看详情
2021年北京市GCP证书线上培训班来啦!具备GCP证书发放资质,效力等同高研院,支持备案。GCP证书是进入临床行业的敲门砖,在CRO公司、医疗机构及各大生产企业应用广泛。目前飞速度已为超过400名有需求的小伙伴,成功发放GCP证书。
查看详情
深圳申请第二类医疗器械注册证的条件《医疗器械注册管理办法》(国家食品药品监督管理总局令第四号)在中华人民共和国境内销售、使用的医疗器械,应当按照本办法的规定申请注册
查看详情
常有人问?第二类医疗器械经营申请如何备案?飞速度许可证办理网为申请人整理如下:上海第二类医疗器械如何备案?1、《医疗器械监督管理条例》(中华人民共和国国务院令第680号,2017
查看详情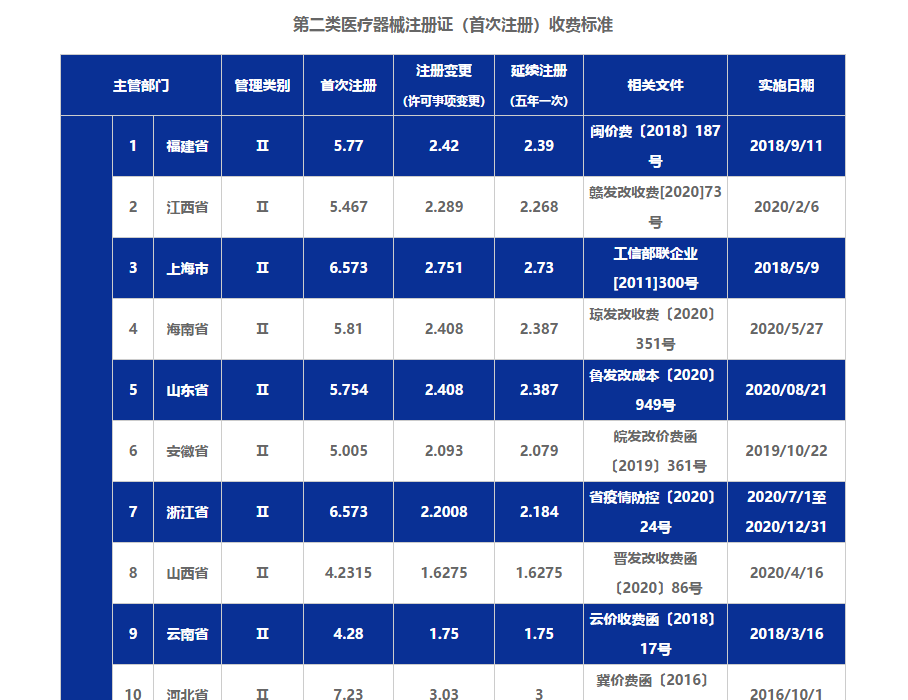
二类医疗器械注册不是一个比较简单的事情,从文件、场地、人员等元素的费用考量外,还需要知道代办费用和首次注册收费费用。那么这两种收费情况都是怎么样的,一起随飞速度来看看
查看详情






