24小时服务热线:19103801095
NEWS CENTER
Recommend case
contact us

我国药物临床试验机构发展开始向备案制过渡,这意味着从监管层面对试验要求的进一步提高,而药品临床试验机构(Good Clinical Practice,GCP)在临床试验的规范过程中发挥着非
查看详情
3D打印椎间融合器技术审评要点(2022年第13号)旨在指导医疗器械注册申请人对3D打印椎间融合器产品注册申报资料的准备及撰写,同时也为技术审评部门审评注册申报资料提供参考。
查看详情
一次性使用病毒采样管通常由拭子和/或含保存液的杯、管等组成。非无菌提供。用于样本的收集、运输和储存等。近期问到一次性使用病毒采样管备案?的客户较多,因此,写篇文章,为大家做个科普。
查看详情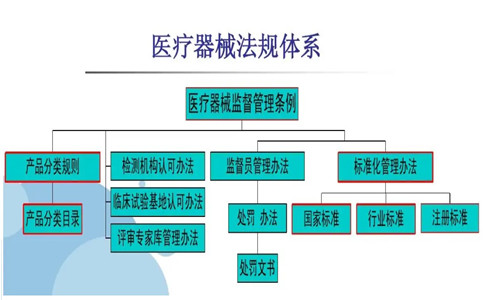
近日有客户咨询到有关医疗器械法规标准体系相关事项,所以,以3D打印椎间融合器这个产品为例,为大家科普医疗器械注册产品适用医疗器械法规标准体系。
查看详情
美国食品药品监督管理局时隔16年第三次修订医疗器械软件申报指南,于2021年11月发布了医疗器械软件功能申报指南草案公开征求意见,拟取代2005年5月发布的医疗器械软件申报
查看详情
近日,威尔史密斯是个话题,不由得想起很多年前的大学时期,看过的他主演的有关人工智能的电影,由此想到了人工智能在医疗器械行业的逐渐应用。因此,为大家带来科普文章,有关人工智能医疗器械监管研究进展。
查看详情
体外诊断仪器通常是指在体外对人体样本进行检测或处理,从而获取临床诊断信息,进而判断疾病或机体功能的一类医疗器械。体外诊断仪器一般需要配合其他仪器或者试剂盒使用,从而达到体外诊断(IVD,In Vitro Diagnosis)的目的。随着体外诊断行业的飞速发展,体外诊断仪器也成为当今医疗手段的重要组成部分,体外
查看详情
2022年2月11日, 国家药品监督管理局发布并实施新版境内第三类医疗器械注册质量管理体系核查工作程序(药监综械注〔2022〕13号),原境内第三类医疗器械注册质量管理体系核查工作程序暂行版(食药监械管〔2015〕63号)同时废止。 新版境内第三类医疗器械注册质量管理体系核查工作程序依据新版《医疗器械监督管理条例》(国务院739号令)及配套的注册与
查看详情






