24小时服务热线:19103801095
NEWS CENTER
Recommend case
contact us
虽然Medical Devices Regulation(MDR)(2017/745/ EU)已经明确将替代Medical Devices Directive (93/42/EEC)(MDD)和 Active Implantable Medical Devices Directive (90/385/EEC)(AIMDD),但是对于新法规的一些具体的内容,尤其是MDR里面那些比较新的要求,还是多少让人摸不着头脑。并且,已经获得MDD认证的器械虽仍可在市场上合法销售,但在2024年5月26日之后,市场上将只允许MDR认证后贴有CE标志的器械准入,那么,在向MDR过渡的这段时间内,我们应该做哪些准备呢?

上市后临床跟踪
今天我们来聊一下上市后临床跟踪,也就是Post-Market Clinical Follow-up (PMCF), 它是上市后监督(PMS)中重要的一部分。MDR的延期,对制造商来说意味着将有更多的时间来收集临床数据,而不是意味着PMCF活动被推迟,这些额外的时间为在2019年或2020年获得认证的器械提供了一个收集临床证据的机会。
在MDD里,对临床数据的质量的定义是含蓄而模糊的,而MDR却十分明确,声明必须由与实际器械(或狭义的等同器械)相关的数据支持,并且必须详细而具体地说明数据质量。因此,那个仅依靠旧文献来demonstrate compliance的时代即将过去,在计划向MDR过渡的时期,调查现有临床数据的可用性和质量是很有必要的。

PMCF Studies
在MDR中,Post-Market Clinical Follow-up(PMCF)的定义是:a continuous process that updates the clinical evaluation and shall be addressed in the manufacturer's post-market surveillance plan(Annex XIV, part B)
而在MEDDEV中,对Post-Market Clinical Follow-up(PMCF)Study是这样定义的,a study carried out following the CE marking of a device and intended to answer specific questions relating to clinical safety or performance(i.e., residual risk)of a device when used in accordance with its approved labelling.
首先,PMCF是一项长期而持续的活动,旨在于方便制造商检查器械的预期寿命内产品的临床安全性和性能,确认先前确定的风险是否可接受,并且及时地发现器械在应用中的新风险(即使是在产品已经上市了数年之后)。其次,制造商通过系统的PMCF研究,在产品的日常使用中主动获取临床数据且对该数据进行及时的评估,纳入研究的器械应当是已经CE-marked的且按照其预期用途使用的器械。
在MEDDEV 2.12/2 rev.2中,详细地阐述了PMCF研究的必要性。该指南要求通过系统的PMCF研究,调查和评估产品上市后的剩余风险。诚然,目前还没有出台法规明确哪些情况需要或不需要做PMCF研究,但在MEDDEV 2.12-2,REV.2中,有提到,PMCF适用于那些基于同产品对比的CE认证(即制造商通过与类似器械的实质等同来证明符合相关基本要求,并且在CE标记之前没有提供该设备本身的长期临床安全和性能数据来说明其适应症的),另外,以下情况也是有可能做PMCF研究的:
1、创新型: 在器械的设计、材料、物质、操作原理、技术或医疗适应症方面是新颖的;
2、产品或预期用途的重大变化(已完成上市前临床评估和再认证的;
3、产品相关风险高;
4、高危解剖部位;
5、高危目标人群(如儿童、老人);
6、疾病的严重程度/治疗挑战;
7、出现有关安全或性能的新信息等。
若制造商评估其自己的产品没有做PMCF研究的必要,或可以在其PMS计划中做出阐述。具体的变化还依据法规动态。
虽然MEDDEVs不被认为是官方文件,也将在MDR过渡时期结束后被取代,但这个MEDDEV在很大程度上已经被MDR吸收。它是目前比较值得参考的指南了。

PMCF Plan
关于如何执行PMCF,你需要制定一份PMCF计划,在PMCF计划中写明了每年计划的活动,然后依据这些活动来执行PMCF。根据MDR, PMCF计划是特定于产品的,因此需要为每个产品准备单独的计划。所有类别的医疗器械都需要有这一计划,其范围将因产品的复杂性和不同风险而有所不同。
根据MDR附录XIV Part B,执行PMCF应当遵循PMCF计划中规定并记录的方法。
PMCF计划至少应包含:
1、待采用的PMCF的通用方法和流程,如收集所获得的临床经验和使用者反馈,筛选科学文献和临床数据的其他来源;
2、待采用的PMCF的专用方法和流程,如对相应注册人员或PMCF研究的评估;
3、 1和2中所述的方法和流程的适当理由;
4、对第 4 节中所述的临床评价报告相关部分和附录I第3节中所述的风险管理的引用;
5、需通过PMCF完成的具体目标;
6、对等同或类似器械的相关临床数据的评估;
7、参考制造商使用的任何相关CS、协调标准和PMCF相关指南;
8、对由制造商执行的 PMCF活动(如对 PMCF 数据的分析和报告)的详细且充分合理的时间安排。
Tip:
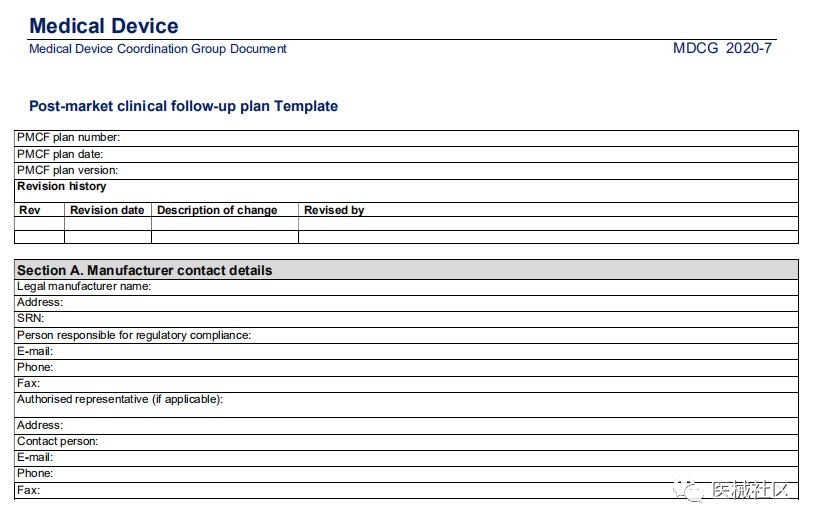
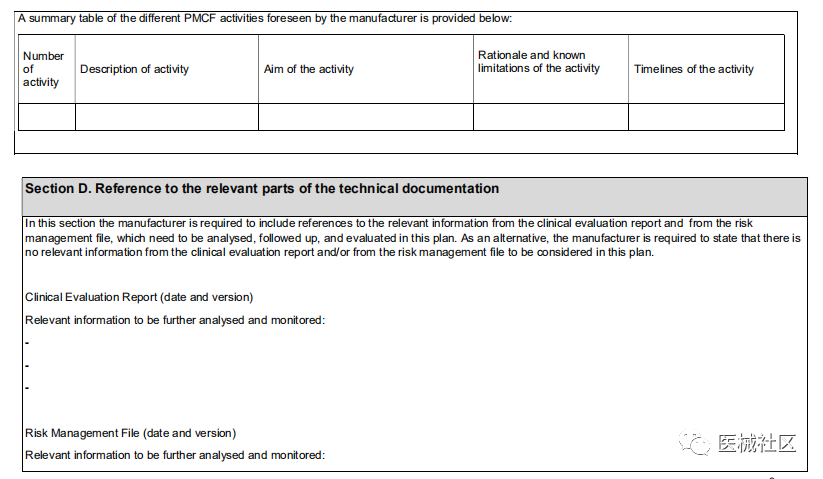
2020年4月,MDCG发布了编号为MDCG 2020-7的指南文件,标题是PMCF计划模板-给医疗器械制造商和公告机构的指南。模板如下:







Reference:
-Medical Devices Regulation(MDR)(2017/745/ EU)
-MEDDEVs
-Post-Market Clinical Follow-up Studies White Paper A logical start to compliance under the new Medical Devices Regulation 2017/745/EU
来源:医械社区

站点声明:
本网站所提供的信息仅供参考之用,并不代表本网赞同其观点,也不代表本网对其真实性负责。图片版权归原作者所有,如有侵权请联系我们,我们立刻删除。如有关于作品内容、版权或其它问题请于作品发表后的30日内与本站联系,本网将迅速给您回应并做相关处理。
北京飞速度医疗科技有限公司专注于医疗器械、诊断试剂产品政策与法规规事务服务,提供产品注册申报代理、临床合同(CRO)研究、产品研发、GMP质量辅导等方面的技术外包服务。











