24小时服务热线:19103801095
NEWS CENTER
Recommend case
contact us
欧盟区是各医疗器械制造商及体外诊断(IVD)产品生产商的核心目标市场之一,而只有满足当地法规的要求,产品才能获得欧盟区市场准入资格。IVDR新法规对IVD生产商来说,提高了欧盟市场准入门槛,并且对其监管更加严格。相信意欲进军欧盟市场的生产商,都需要开展紧锣密鼓的法规跟踪与实施工作。

2017年5月5日欧盟体外诊断医疗器械法规(IVDR)正式发布,并于2017年5月25日正式生效,2022年5月26 日实施。自实施之日起,IVDR 将取代原欧盟体外诊断设备指令(IVDD)。对于IVD产品生产商,新法规带来变化有哪些?对临床证据的要求又有何变化?
IVDR按照风险等级将IVD产品分为四类:Class A(风险最低)、Class B、Class C和Class D(风险最高)。IVDR法规规定B,C,D类的体外诊断试剂都需要由公告机构(Notified Body,NB)认证。涉及公告机构介入的产品数量从IVDD监管体系下的10%~20%增加至80%~90%。
公告机构介入量的增加,意味着绝大多数的体外诊断设备,在欧盟区的市场准入将要告别原先“自我声明”的形式,取而代之的将是一个实质性的注册过程。因此,体外诊断试剂的厂商应先确定产品在IVDR中的分类,并尽早同公告机构取得联系。
IVDR公告机构查询网址(打开较慢,耐心等待):https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=35
随着IVDR法规的实施,IVD产品需要的临床证据也将增加。可下载“Clinical Evidence Requirements for CE certification under the in-vitrio Diagnostic Regulation in the European Union”文件详细参阅。您可以进入下方网址,下载文件。
网址:https://www.medtecheurope.org/resource-library/clinical-evidence-requirements-for-ce-certification-under-the-in-vitro-diagnostic-regulation-in-the-european-union/
此外,2020年4月份委员会小组起草的关于新冠检测方法和设备的性能要求文件-“Current performance of COVID-19 test methods and devices and proposed performance criteria” 也具有重要参考价值。点击前面文件名可直接下载文件。
新法规强调对供应链的格外监督,包括公告机构对关键供应商和分包商的突击审核。此外,新法规规定了进口商、分销商和授权代表的监管作用以及对他们的要求。制造商必须为其产品投保责任保险;但是,进口商有责任验证保险是否充分,否则进口商必须购买额外的保险。
除了法定制造商,授权代表必须指定专人负责其组织内部的法规遵从工作。这个专人必须在IVD领域受到过适当的教育以及具有IVD领域的经验。而且,他还必须对技术文档负责,确保符合性声明、性能评估和监测要求及时更新。
MedTech Europe拟定了一份流程图,可以从中一览IVDR法规下的新要求。
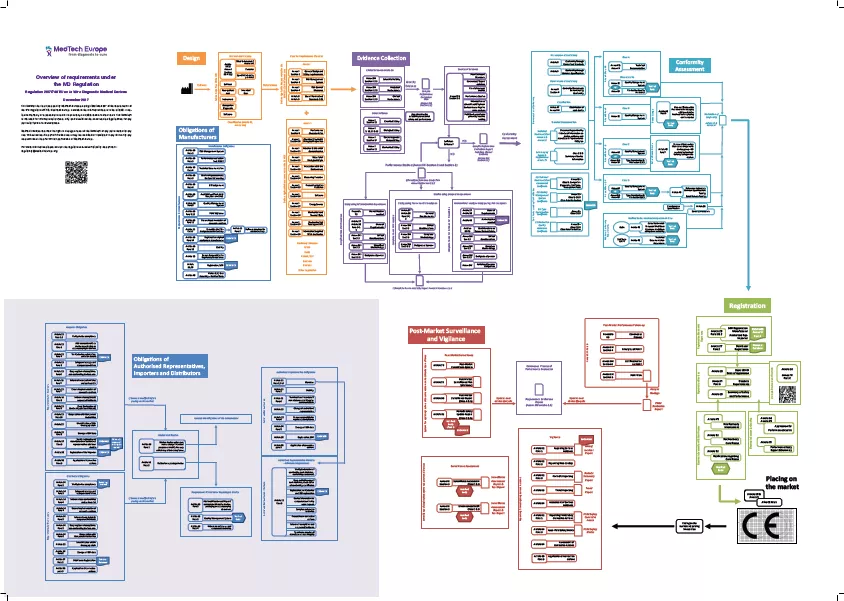
完整流程图,点击下载。

站点声明:
本网站所提供的信息仅供参考之用,并不代表本网赞同其观点,也不代表本网对其真实性负责。图片版权归原作者所有,如有侵权请联系我们,我们立刻删除。如有关于作品内容、版权或其它问题请于作品发表后的30日内与本站联系,本网将迅速给您回应并做相关处理。
北京飞速度医疗科技有限公司专注于医疗器械、诊断试剂产品政策与法规规事务服务,提供产品注册申报代理、临床合同(CRO)研究、产品研发、GMP质量辅导等方面的技术外包服务。











