24小时服务热线:19103801095
NEWS CENTER
Recommend case
contact us

上海市药品监督管理局关于发布《上海市第一类医疗器械备案工作指南》的通告
2020年 第20号
为进一步指导各区市场监督管理局和备案人依法规范开展第一类医疗器械备案工作,上海市药品监督管理局依据《医疗器械监督管理条例》等相关法规规章文件,结合年内开展的第一类医疗器械备案管理工作考核评估情况,组织制定了《上海市第一类医疗器械备案工作指南》,现予发布。特此通告。
上海市药品监督管理局
2020年11月19日
(公开范围:主动公开)
上海市第一类医疗器械备案工作指南
(2020年制订)
依据《医疗器械监督管理条例》等相关法规规章文件,结合年内开展的第一类医疗器械备案管理工作考核评估情况,制定《上海市第一类医疗器械备案工作指南》。
本工作指南旨在规范本市第一类医疗器械备案工作,分为第一类医疗器械产品备案、第一类医疗器械生产备案两个部分,突出重点为各区市场监督管理局开展相关备案工作提供参考。
本工作指南不作为法规强制执行,但随着法规的不断完善,将适时对相关内容予以调整。
第一部分 第一类医疗器械产品备案
一、备案依据
1.《医疗器械监督管理条例》
2.《医疗器械注册管理办法》
3.《体外诊断试剂注册管理办法》
4.《原国家食品药品监督管理总局关于第一类医疗器械备案有关事项的公告》(2014第26号)
5.《原国家食品药品监督管理总局关于实施第一类医疗器械备案有关事项的通知》(食药监办械管〔2014〕174号,以下简称“174号文”)
二、适用范围
适用于上海市第一类医疗器械产品备案(包括首次备案、备案变更、备案凭证遗失补办、取消备案号四种办理情形)。
三、审查重点
(一)总体要求
1.自2014年6月1日起,国家对第一类医疗器械实行产品备案管理,属于目录化管理。备案人作出相关承诺,对备案资料的真实性、完整性、合规性负责;各区市场监管局对备案人提交的符合形式审查要求的备案资料存档备查。
2.2017年2月1日起,备案人向生产地所在区市场监管局提交备案资料。2017年2月1日前,已按原规定在备案人住所地区市场监管局完成产品备案的,其相关备案凭证继续有效。
3.2017年2月1日前已备案产品,其备案资料载明的事项发生变化的,如仍属于备案人住所地与生产地跨区的情形,备案人应向原备案部门提出取消产品备案,并按更新后的内容重新向备案人生产地所在区市场监管局提出备案(不强制要求先取消产品备案)。
4.递交资料形式和内容应符合“一网通办”平台中相关办事指南的要求。
(二)“首次备案”形式审查关注重
1.明确备案依据
备案人在符合性声明中应注明确切的产品分类依据,以下为原则性的要求:
(1)2014年国家局下发的《第一类医疗器械目录》(需注明所属子目录以及相应序号)
(2)《医疗器械分类目录》(2017版)(需注明所属子目录、序号以及二级产品类别)
(3)《体外诊断试剂分类子目录》(2013版)(需注明序号以及产品类别)
(4)《关于过敏源类等体外诊断试剂产品属性及类别调整的通告》(2017年第226号)(需注明相应附件名称以及序号)
(5)国家局于2014年6月1日之后下发的分类界定文件(需注明具体文号、文件名、引用的具体内容)
(6)“医疗器械分类界定信息系统”平台中的《分类界定告知书》(需注明受理号、告知号以及《分类界定告知书》中相关产品信息的具体内容)
(7)如有其他相关依据也要注明具体出处和内容。
举例1:
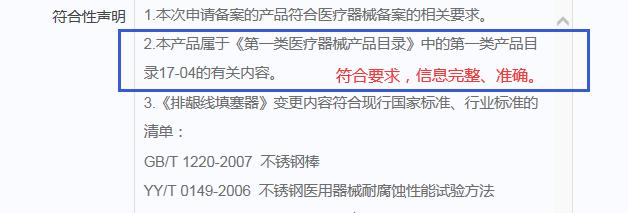
举例2:
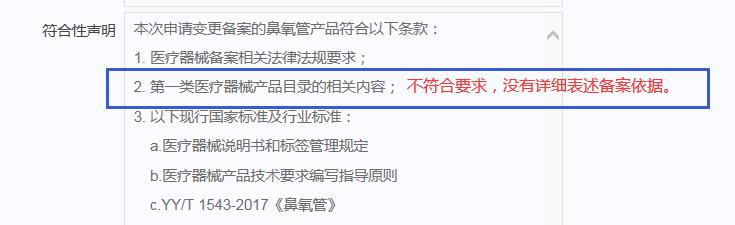
2.规范产品名称
1)原国家食品药品监督管理总局办公厅《关于实施第一类医疗器械备案有关事项的通知》(食药监办械管〔2014〕174号)明确“根据所属类别,应直接使用目录中“品名举例”所列举的名称”。
2)《第一类医疗器械目录》《医疗器械分类目录》等目录中不包含组合包类产品;《体外诊断试剂分类子目录》(2013版)中包含的第一类体外诊断试剂品种较少,因此,可以按第一类医疗器械管理的组合包类产品以及部分体外诊断试剂产品名称的确定,应符合174号文的相关规定。
3)产品名称不应包含“产品的型号或规格”“人名、企业名、品牌名、商标名或其它类似的名称”“最佳、最新、唯一、精确、速效等绝对化或排他性的词语”“明示或者暗示对某种疾病具有治疗作用的词语,或含有表示功效、说明有效率和治愈率的断言或者保证”、“明示或暗示包治百病、适应所有症状或者夸大适应症的内容,或含有美容、保健等宣传性内容”“未经科学发现证明或临床结果证明,或虚无、假设的概念性名称”。
3.规范型号/规格的表述方式
应清晰、准确表述产品的全部型号/规格,型号/规格可以以使用部位、尺寸、装量等形式表述,例如:额面型、足底型、手心型、腋窝型;5cm*10cm;5片/盒、5瓶/盒、50g/支等。
对于医用冷敷贴、医用冷敷眼罩、冷敷凝胶、液体敷料、喷剂敷料、伤口护理软膏、液体伤口敷料等产品,说明书、内外包装标签、产品宣传等信息载体中,关于产品型号/规格(包括产品名称、产品描述、预期用途/适用范围等)的表述内容均不应体现超出备案产品描述以及预期用途范畴的表述内容,例如:修护(修复)型、精华型、消肿止痛型、缓解过敏型、抑制色素沉着型、消痘型、抗炎型等。
举例1:
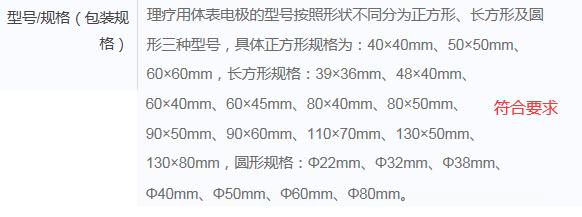
举例2:
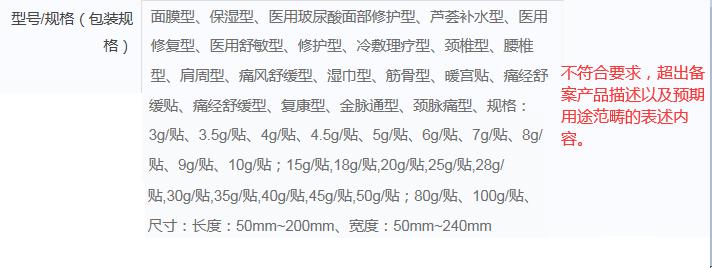
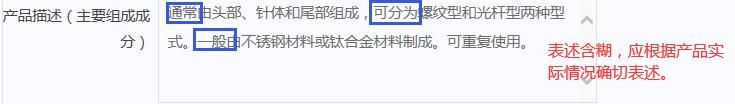
4.规范产品描述
1)相关目录“产品描述”栏目,如存在“通常”、“一般”等表述内容,是指对纳入相应栏目产品的基本描述,备案人应针对具体产品准确表述,体现产品技术特点。
2)相关目录中有“除外”和特别注明情形的,应在备案的产品描述中应予以说明,具体要求详见174号文。
3)不应超出相关目录“产品描述”内容的范围。
4)可以按第一类医疗器械管理的组合包类产品以及部分体外诊断试剂的产品描述,应符合174号文的相关规定。
举例:
5.规范预期用途
1)“预期用途”的基本内容应与目录中的相应内容一致。
2)相关目录“预期用途”栏目,如存在“通常”“一般”等表述内容,是指对纳入相应栏目产品预期用途的基本描述,备案人应针对具体产品准确表述,不应照搬照抄相关内容。
3)可以按第一类医疗器械管理的组合包类产品以及部分体外诊断试剂的预期用途,应符合174号文的相关规定。
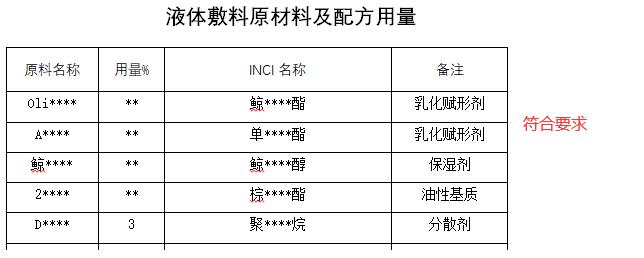
6.生产制造信息
对于第一类敷料敷贴类产品(包括医用冷敷贴、医用降温贴、医用退热贴、医用冰袋、医用冰垫、医用冰帽、医用冷敷头带、医用冷敷眼罩、冷敷凝胶;液体敷料、喷剂敷料、伤口护理软膏、液体伤口敷料;创口贴等),备案人应在备案资料“生产制造信息”中提交所有添加成分以及含量信息,并如实表述所有成分的作用。
审核过程中应重点关注以下内容:
1)申请备案产品配方中添加的相关成分未被《中华人民共和国药典》收载。
2)申请备案产品配方中添加的相关成分,即使未被《中华人民共和国药典》收载,例如天然植物或其提取物、防腐剂、抑菌剂、透明质酸钠、胶原蛋白等,同样不具有药理学作用。
3)液体敷料、喷剂敷料、伤口护理软膏、液体伤口敷料、创口贴的产品配方中添加的相关成分不可被人体吸收。
4)医用冷敷贴、医用降温贴、医用退热贴、医用冰袋、医用冰垫、医用冰帽、医用冷敷头带、医用冷敷眼罩、冷敷凝胶的产品配方中,相关降温物质不得发挥免疫学或者代谢作用。
若备案人认为产品配方中添加的相关成分不具有药理学作用、不可被人体吸收;其中降温物质不发挥药理学、免疫学或者代谢作用,应提供相应的证明材料。备案部门与备案人就此无法达成一致意见时,可建议备案人按规定申请分类界定。
举例:
7.关于产品实物照片
《医疗器械注册管理办法》第十一条规定“申请人或者备案人申请注册或者办理备案,应当遵循医疗器械安全有效基本要求,保证研制过程规范,所有数据真实、完整和可溯源。”因此,办理第一类医疗器械产品备案的前提条件并非仅仅完成备案资料的编撰工作,备案人应已完成样品的研发和生产。备案人应在备案资料中递交拆除所有内、外包装后的样品实物照片,以及内外包装实样照片。
8.关于接触人体的有源医疗器械
根据医疗器械分类规则判定表,接触人体的有源医疗器械一般不属于第一类医疗器械,若相关目录未明示可以按照第一类医疗器械管理,且备案部门与备案人无法达成一致意见时,可建议备案人按规定申请分类界定。
9.关于使用前需经灭菌处理的非无菌第一类医疗器械
备案人应在产品说明书中明确灭菌方法,并对灭菌方法进行验证确认,相关验证确认资料可作为备案资料递交,也可由备案人自行保存备查。
10.线上线下递交资料一致性
本市第一类医疗器械产品备案已实现网上申请、网上受理、网上办结,对于仍要求备案人递交纸质备案资料的,备案人应确保线上线下递交资料一致。
11.关于第一类医疗器械委托生产
第一类医疗器械备案目前不适用实施医疗器械注册人制度(即备案人自身无生产能力,委托具备相应生产能力的企业生产产品和样品)。
(三)“备案变更”形式审查关注重点
1.明确备案依据
备案人在符合性声明中应注明确切的产品分类依据,表述内容的原则性要求,同首次备案,同时需要关注相关变更内容不影响产品类别的改变。
2.规范产品名称
如涉及产品名称的变更,需要关注相关变更内容不影响产品类别的改变,同时变更后的产品名称直接使用目录中“品名举例”所列举的名称(除外情形:可以按第一类医疗器械管理的组合包类产品以及部分体外诊断试剂产品名称的确定,应符合174号文的相关规定)。
3.规范型号/规格的表述方式
对于第一类敷料敷贴类产品(包括医用冷敷贴、医用冷敷眼罩、冷敷凝胶、液体敷料、喷剂敷料、伤口护理软膏、液体伤口敷料等),变更后的型号/规格表述要求,同首次备案。
4.规范产品描述
变更后产品描述的表述要求,同首次备案。
5.规范预期用途
变更后预期用途的表述要求,同首次备案。
6.准确表述备案变更内容
在备案变更申请表“变更情况”栏目,简明扼要具体表述变更内容,避免表述过于笼统引起公示内容含糊不清。
举例:变更情况仅填写“有效期变更”过于简单。可填写变更后内容“有效期变更为×××”,或者填写变更前后比对内容“有效期由×××变更为×××”。
7.关于第一类敷料敷贴类产品变更所含成分
如变更第一类敷料敷贴类产品的成分,备案人应在备案资料“企业认为需要申报的其他文件材料资料”中,提交变更后成分以及含量信息,并如实表述所有成分的作用。
审核关注点同首次备案。
8.线上线下递交资料一致性
本市第一类医疗器械产品备案已实现网上申请、网上受理、网上办结,对于仍要求备案人递交纸质备案资料的,备案人应确保线上线下递交资料一致。
(四)“备案凭证遗失补办”、“取消备案号”形式审查关注重点
备案表填写完整不留白,并通过法人一证通签章。
第二部分 第一类医疗器械生产备案
一、备案依据
1.《医疗器械监督管理条例》
2.《医疗器械生产监督管理办法》
3.《原国家食品药品监督管理总局关于医疗器械生产经营备案有关事宜的公告》(2014第25号)
4.《原国家食品药品监管总局关于印发医疗器械生产企业分类分级监督管理规定的通知》(食药监械监〔2014〕234号)
二、适用范围
适用于第一类医疗器械生产备案(包括首次备案、备案变更、备案凭证补办、备案凭证注销、医疗器械委托生产备案等五种办理情形)。
三、审查重点
(一)首次备案
1.生产场地
企业应提供生产场地的不动产权证(房地产权证)和租赁协议(如申请人为房地产权人,则仅需提交产权证),产权证由不动产登记部门核发,相关材料应能明示不动产登记信息中的房屋类型、部位等信息。审核应重点关注生产区域、检验场所与其产品生产规模、品种相适应性。
2.设备文件
企业应逐一填写主要生产设备、主要检验设备目录,质量手册和程序文件目录。备案部门应在完成备案3个月后,对企业申报的生产设备、检验设备、质量手册、程序文件等相关内容进行核实。
3.工艺流程图
企业提供的产品工艺流程图中,应明确标识所生产产品的关键工序和特殊过程。
4.产品相关信息
生产备案的首次备案和涉及产品变化的变更备案,需要提供产品技术要求、产品备案凭证等产品相关技术资料,上述资料应提供备案部门盖章的最终版本。
(二)备案变更
1.生产地址变更
(1)跨区增设生产地址,各区市场监管局应通过备案前信息互通和备案后跨区监管切实加强监管协同。同一企业法人主体对应一个第一类医疗器械生产备案,可通过变更备案的方式新增产品、新增生产地址。生产备案凭证编号以首次备案为准,变更备案不对备案凭证编号进行变化。涉及跨区域生产的第一类医疗器械,备案部门可在备案凭证生产产品列表一栏中进行备注。
(2)跨区生产地址搬迁,备案人应向原备案部门提出注销生产备案凭证,并重新向备案人生产地所在区市场监督管理局提出生产备案。
2.生产范围、产品登记表变更
企业取得新的产品备案或减少产品备案后,应向该产品生产地址所在辖区提出生产备案变更。
(三)备案注销
区市场监管局发现不符合一类生产备案条件的,依法予以注销。
(1)一类医疗器械生产企业不具备原生产备案条件,或者与备案信息不符,且无法取得联系的,经备案部门公示后,依法注销第一类医疗器械生产备案信息,并向社会公告;
(2)在备案后核查中,发现一类医疗器械生产企业提供虚假备案资料的,依法予以查处,并撤销企业医疗器械生产备案凭证;
(3)在备案后核查中,发现一类医疗器械生产企业质量管理体系存在缺陷,依法要求企业进行整改;情节严重的,可采取责令整改等措施,直至注销一类生产备案。
各区市场监管局应每年度定期将相关信息报送市药品监管局。
(四)委托生产备案
委托方在同一时期只能将同一医疗器械产品委托一家医疗器械生产企业(绝对控股企业除外)进行生产。受托方办理备案后,备案凭证中的受托生产产品应当注明“受托生产”字样和受托生产期限。委托生产医疗器械的说明书、标签除应当符合有关规定外,还应当标明受托方的企业名称、住所、生产地址、生产许可证编号或者生产备案凭证编号。委托生产终止后30天内,委托方和受托方应当向所在区市场监管局及时报告。
(五)备案补办
目前一类生产备案办事项目已实现“全程网办”。备案部门通过“一网通办”系统推送电子备案凭证,原则上不再打印纸质凭证。企业如需纸质备案凭证,可自行打印或向备案部门提出申请。对于存量的纸质备案凭证补办,仍按照备案补办事项的办事指南执行。
上海市第一类医疗器械备案工作指南2020年制订.docx(160.22 KB)

站点声明:
本网站所提供的信息仅供参考之用,并不代表本网赞同其观点,也不代表本网对其真实性负责。图片版权归原作者所有,如有侵权请联系我们,我们立刻删除。如有关于作品内容、版权或其它问题请于作品发表后的30日内与本站联系,本网将迅速给您回应并做相关处理。
北京飞速度医疗科技有限公司专注于医疗器械、诊断试剂产品政策与法规规事务服务,提供产品注册申报代理、临床合同(CRO)研究、产品研发、GMP质量辅导等方面的技术外包服务。











