24小时服务热线:19103801095
NEWS CENTER
Recommend case
contact us

ISO9001认证指的是质量体系认证,对于企业来说,这个认证是必要的,因为在如今的这个市场上,人们对于产品的质量要求越来越高,如果没有ISO9001认证的话,企业的产品甚至会无人问津。本文来解决两个企业关注的高热度问题,即ISO9001认证硬性条件和通过ISO9001认证企业能享受到哪些好处.......
查看详情
ISO9001认证是质量管理体系的认证,对于很多公司而言,进行认证咨询有利于提升管理、开拓市场。企业的质量不仅仅是某个活动的工作质量,还可以是指涉及人的素质、设备的能力、管理水平的体系运行的质量。下面小编来介绍一下申请iso9001认证需要多少钱?
查看详情
ISO9001认证体系,可以说是对于企业的一个质量的保障,是很多企业都需要的一个认证,只有获得这个认证,才代表着你的产品质量得到了市场的肯定。能够更好的进行市场的交易。下面小编来介绍一下iso9001认证步骤和流程。
查看详情
ISO9001在大家心里,只感觉是一个很权威的认证。平时我们生活当中会看到喝的饮料还有路过的工厂、以及一些其他的商品上会标记说获得ISO 9001认证?这些认证是由什么机构来
查看详情
在做ISO9001认证的时候也是需要相应的条件的,首先公司的营业时间必须满三个月,还有就是公司必须做过咨询。那么接下来我们一起来看看iso9001认证申请条件和申请流程。
查看详情
申请人提交材料目录:资料编号(一) 境内第三类医疗器械注册申请表;
资料编号(二) 医疗器械生产企业资格证明;
资料编号(三) 产品技术报告;
资料编号(四) 安全风险分析报告;
资料编

iso9001质量体系认证是什么意思?iso9001是指什么?ISO9000是被全球认可的质量管理体系标准。ISO9001:2000是国际标准化组织融合现代管理学最新的理念精华,推出的最新质量管理体系标准,更加适用于各种类型、各种行业的组织。ISO9001:2000为组织提供了一种切实可行的的方法,以体系化模式来管理组织的质量活动,并将“以顾客为中心”的理念贯穿到标准的每一元素中去,使产品或服务可持续地符合顾客的期望,从而拥有持续满意的顾客。
查看详情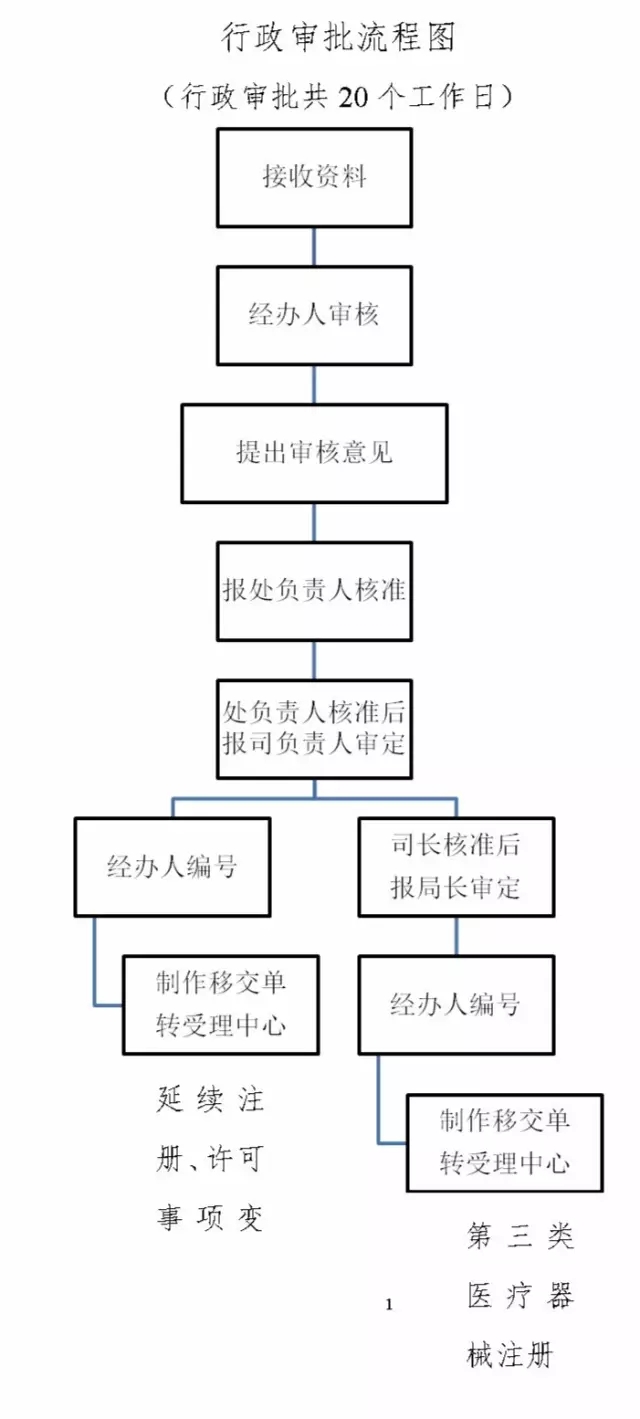
国产第三类医疗器械首次注册审批流程国产第三类医疗器械首次注册审批一、适用范围 本指南适用于的申请和办理国产第三类医疗器械首次注册。二、项目信息 (一)项目名称
查看详情






