24小时服务热线:19103801095
NEWS CENTER
Recommend case
contact us
二尖瓣缘对缘修复技术(edge-to-edge repair)在1991年由意大利学者Alfier提出,该技术是指将二尖瓣进行缘对缘缝合,从而使二尖瓣双孔化(A2/P2区)或者关闭A1/P1或A3/P3区对合缘达到治疗反流的目的(见图1、图2)。而二尖瓣夹合器系统则是基于外科二尖瓣缘对缘修复技术,利用机械夹合的原理,通过经导管介入手术植入人体后,二尖瓣夹合器的上夹、下夹夹持住二尖瓣瓣叶,成“V”形的上下夹两臂向中线靠拢,同时带动所夹合的组织向中线靠拢,收入闭合环后即锁定,然后释放夹合器。植入二尖瓣夹合器后,可减少二尖瓣的反流,达到治疗反流的效果。

一、FDA已上市经导管二尖瓣夹系统临床评价简介
(一)经导管二尖瓣夹及可操控导引导管MitraClip System
MitraClip System是首个在FDA获批的二尖瓣夹合器系统,在开展了EVEREST I Feasibility可行性研究后,2005年开始了EVEREST II RCT(以下简称RCT)研究。该临床研究的对照组为外科手术,受试者为预期接受二尖瓣外科手术的人群。试验过程中,部分重度二尖瓣反流患者经医生判断由于外科手术风险过高而无法入组RCT研究,因此2007年该项目补充开展了EVEREST II HRR(以下简称HRR)单臂临床研究,并入组二尖瓣外科手术高风险人群,RCT和HRR同步筛选受试者。RCT和HRR完成入组后, Continued Access Registry REALISM于2009年启动,该研究包含两个队列:外科手术高风险队列(以下简称HR)和非外科手术高风险队列,该研究可供医生和患者在MitraClip System注册申报期间继续使用该产品的同时,申办方也可继续收集安全性和有效性数据。
根据FDA公开的审评信息,尽管RCT研究证实MitraClip System可以被安全植入并降低大部分患者的二尖瓣反流,但有效性不及外科手术(具体临床试验结果见表1),因此该临床研究不足以支持MitraClip System在手术候选人群中使用。
表1 EVEREST II RCT临床试验结果总结
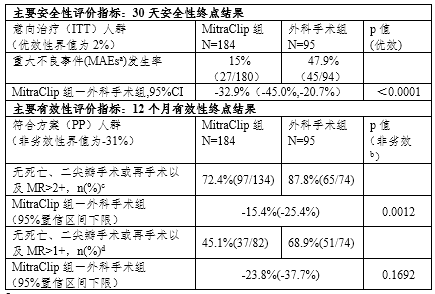
a. MAEs是包括死亡、心肌梗死(MI)、修复或置换手术失败而导致的再手术、不良事件导致的非计划心血管手术、中风、肾功能衰竭、深部创口感染、连续48小时通气、胃肠道(GI)并发症导致的手术、新发的持续性房颤、败血症以及输注2个及以上单位的血液等临床事件的复合指标。
b. 此处p值是基于非劣效界值为-31%计算出来的,然而由于非劣效界值过大,该结果不意味着存在非劣效性结论。
c. 申办方雅培预设的终点
d. FDA预设的终点
申请人通过与FDA和临床专家探讨,MitraClip System在2011年4月缩窄了申报范围,只针对因外科手术风险过高而无法接受治疗的退行性二尖瓣反流(Degenerative Mitral Regurgitation,DMR)和功能性二尖瓣反流(Functional Mitral Regurgitation,FMR)患者。HRR和REALISM HR(REALISM非外科手术高风险队列于2011年9月暂停入组)两个单臂临床研究针对这部分人群开展,但是存在局限性,包括:二尖瓣反流病因的异质性、数据合并、事后设置对照组(杜克大学医学中心数据库中65例外科手术高风险DMR患者)、事后分析、数据可靠性以及难以定义受试者的手术风险。
在2013年3月20日的专家咨询会上,与会专家认为EVEREST II HRR和REALISM HR研究中351名患者的临床数据合理地证明了MitraClip System在外科手术高风险人群中的使用是安全的。然而,由于上述的局限性,尤其是受试者手术风险定义和二尖瓣反流病因的异质性,大部分专家咨询小组成员无法得出有效性的结论。会后,FDA与申请人共同决定使用HRR和REALISM HR入组的外科手术高风险DMR患者来评价MitraClip System的风险受益,因为经导管二尖瓣缘对缘修复术对DMR患者减少二尖瓣反流的价值更为明确。DMR是一种瓣膜出现的“机械故障”,目前尚无有效药物手段来降低DMR导致的二尖瓣反流,只能用“机械”的方法来矫正。但对于FMR来说,最佳药物治疗有一定受益,因为FMR继发于左心室功能障碍,而左心室功能障碍可以通过药物治疗、血运重建和/或心脏再同步治疗得到改善。因此,MitraClip System在FMR中的临床受益无法通过现有的单臂研究结果来确定。
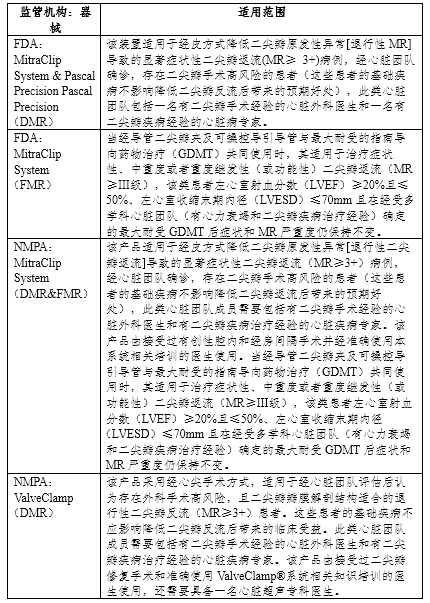
最终,通过对EVEREST II、HRR和REALISM中127例外科手术高风险DMR患者的临床数据进行分析,FDA批准了MitraClip System用于二尖瓣原发性异常(退行性二尖瓣反流)导致的显著症状性二尖瓣反流(MR≥3+)病例,经心脏团队确诊,存在二尖瓣手术高风险的患者(完整适用范围见表2)。
针对FMR,MitraClip System在2012年12月开展了COAPT研究,在美国和加拿大的78个中心招募了614名患有中重度或重度二尖瓣反流,并且在最大药物治疗剂量下仍有症状的心力衰竭患者。受试者被随机分至接受经导管缘对缘修复术联合指南导向药物治疗组(MitraClip System + GDMT)或仅接受指南导向药物治疗(GDMT)组。试验的主要有效性终点是两年内的心衰住院率,主要安全性终点是12个月内无器械相关安全性事件发生的概率。基于COAPT研究,FDA批准了MitraClip System用于使用最大剂量的指南导向药物治疗后仍有症状的中重度或重度继发性二尖瓣反流患者(完整适用范围见表2)。
(二)Pascal Precision 经导管瓣膜修复系统
2022 年 9 月 15 日, Pascal Precision 经导管瓣膜修复系统获得 FDA 批准,用于治疗退行性二尖瓣反流(DMR)患者(完整适用范围见表2)。Pascal Precision的临床研究CLASP IID包含一个随机对照研究和一个单臂登记队列研究。随机对照研究中受试者按2:1分为两组:PASCAL Precision组和MitraClip System组。单臂登记队列研究入组了被认为由于二尖瓣解剖复杂而无法使用MitraClip System但可以使用PASCAL Precision的患者。该产品主要基于随机对照试验批准,该试验主要有效性终点是6个月时MR≤2+的患者比例(非劣效界值为-18%),主要安全性终点是30天内的MAEs发生率(非劣效界值为15%)。CLASP IID预期纳入300名患者进入随机队列,150名患者进入注册队列。随机队列采用贝叶斯自适应设计,当180、210和240名患者分别达到6个月随访时,允许由一名非盲独立统计学家进行三次期中分析。如果在第一次期中分析中,主要安全性和有效性终点的预测成功率大于96.5%,或在其余两次期中分析中大于95.0%,则将提前宣布成功。试验成功后,仍会继续完成招募300名随机队列患者的计划并随访。PASCAL Precision使用第一次期中分析的结果完成了PMA的申报(PASCAL Precision组有117例受试者,MitraClip System组有63例受试者)。
表2 二尖瓣夹合器系统适用范围
二、我国已上市经导管二尖瓣夹系统临床评价简介及建议
在中国, MitraClip System经导管二尖瓣夹及可操控导引导管主要通过提交境外临床试验数据进行临床评价,还补充了来自文献的中国人群、亚太人群的临床经验数据,并于2020年6月15日和2021年4月20日获批(完整适用范围见表1)。
上海捍宇医疗科技股份有限公司的二尖瓣夹系统于2023年8月31日批准上市,该产品通过经心尖介入治疗方式将二尖瓣夹输送至二尖瓣瓣叶处,二尖瓣夹经超声引导定位并夹持住二尖瓣的前叶和后叶,使瓣叶紧密对接,然后操作输送系统释放二尖瓣夹,从而达到降低二尖瓣反流的目的。该产品通过临床试验路径进行临床评价,采用前瞻性、多中心、单组目标值的试验设计,以验证该产品的安全性及有效性。试验共入组 102 例受试者。有效性评价指标包括术后 360 天时产品有效率(无死亡、无二尖瓣瓣膜再次手术发生率,二尖瓣反流(MR)≤2级),术后即刻、术后 30 天、术后180 天产品有效率(无死亡、无二尖瓣瓣膜再次手术发生率,MR≤2 级)。安全性评价指标包括术后即刻、出院前及 30 天、180 天和 360 天收集并评价以下指标:通过超声等记录的夹合部位发生的不良事件、患者 NYHA 心功能分级、实验室检查异常及判断说明等。该临床试验于2019年启动,当时国内尚无同类产品上市,因此无法使用阳性器械进行对照。该产品适用于经心脏团队评估后认为存在外科手术高风险的患者,因此也无法以外科手术作为对照组。
根据现有经导管二尖瓣夹系统的适用范围和设计特征,对该类器械的临床试验设计提出如下建议:
(一)对于适用于经心脏团队评估后认为存在外科手术高风险,且二尖瓣瓣膜解剖结构适合的退行性二尖瓣反流(MR≥3+)患者的二尖瓣夹合器系统,如参考《医疗器械临床试验设计指导原则》有合理理由可选择单组设计时,考虑到单组设计实质是将主要评价指标的试验结果与已有临床数据进行比较,以评价试验器械的有效性/安全性,存在非同期对照偏倚,包括选择偏倚、混杂偏倚、测量偏倚和评价偏倚等,因此申请人在选择单组设计时应重点关注如下内容:
1.试验器械的适用人群、主要评价指标(如观察方法、随访时间、判定标准等)需被充分定义且稳定,并采取充分的偏倚控制措施。需设置恰当的入排标准,入选标准需保障受试人群对预期使用人群的代表性,排除标准旨在尽可能规范受试者的同质性。临床试验的主要有效性终点建议为术后 12个月产品有效率(无死亡、无二尖瓣瓣膜再次手术发生,二尖瓣反流(MR)≤2级),主要安全性终点建议设为30天内的MAEs发生率。次要评价指标建议考虑:术后6个月和12个月的MAEs发生率、术后30天和6个月的产品有效率、NYHA心功能分级、SF36测量的生活质量、6分钟步行测试、左心室功能(LVEF左室射血分数、LVEDV左室舒张末容积、LVESV左室收缩末容积、LVIDs左室收缩末内径、LVIDd左室舒张末内径)和心衰再住院情况等。
2.目标值的设定需依据充分的循证医学证据,全面收集具有高质量水平及相当数量病例的临床研究数据,并进行科学分析(如Meta分析)。由于单组设计可能存在选择偏倚、混杂偏倚等问题,申请人应详细比对临床试验与用于目标值构建的临床研究数据的患者基线数据,以及各评价指标定义的一致性(如MAEs和“有效”的定义)。由于单组目标值试验无法确证试验器械的优效、等效或非劣效,仅能确证试验器械的有效性/安全性达到专业领域内公认的最低标准,因此,确定的目标值需科学、客观,包括靶值和单侧置信区间界限,确保临床试验可基于充分的样本量得出产品安全、有效的结论。
(二)对于适用于使用最大剂量的指南导向药物治疗后仍有症状的中重度或重度继发性二尖瓣反流患者的二尖瓣夹合器系统,申请人需开展随机对照试验,可选取仅接受指南导向药物治疗(GDMT)或阳性器械作为对照组。
参考文献:
[1] 潘翠珍,潘文志,周达新.二尖瓣反流介入治疗的超声心动图评价中国专家共识[J].中国介入心脏病学杂志,2019,27(01):6-12.
[2] W G S,S A V,H D A, et al. Clinical trial design principles and endpoint definitions for transcatheter mitral valve repair and replacement: part 1: clinical trial design principles: A consensus document from the mitral valve academic research consortium.[J]. European heart journal,2015,36(29).
[3] 医疗器械临床试验设计指导原则(2018年第6号)
[4] FDA P100009 SSED
[5] FDA P100009 S028 SSED
[6] FDA P220003 SSED
临床与生物统计一部 供稿
(原创 2023-10-12 CMDE 中国器审)

站点声明:
本网站所提供的信息仅供参考之用,并不代表本网赞同其观点,也不代表本网对其真实性负责。图片版权归原作者所有,如有侵权请联系我们,我们立刻删除。如有关于作品内容、版权或其它问题请于作品发表后的30日内与本站联系,本网将迅速给您回应并做相关处理。
北京飞速度医疗科技有限公司专注于医疗器械、诊断试剂产品政策与法规规事务服务,提供产品注册申报代理、临床合同(CRO)研究、产品研发、GMP质量辅导等方面的技术外包服务。











